行業(yè)動態(tài)
您當(dāng)前的位置 > 行業(yè)動態(tài) > 行業(yè)新聞
印度醫(yī)療器械監(jiān)管重點:藥品和醫(yī)療器械區(qū)分開來
加入日期:2018/6/5 9:16:22 查看人數(shù): 1057 作者:admin
印度醫(yī)療器械市場位居全球前20,同時也是亞洲第四大醫(yī)療器械市場。其市場規(guī)模大約在55億美元左右,預(yù)計復(fù)合年增長率為15%。
印度的醫(yī)療器械市場在過去的二十年中經(jīng)歷了持續(xù)不斷的變革。在1991年的“新經(jīng)濟政策”之前,印度的醫(yī)療器械在國內(nèi)制造業(yè)中占據(jù)著主導(dǎo)地位。之后,它變成了一個以進口產(chǎn)品驅(qū)動的市場。在2006年之前,印度的醫(yī)療設(shè)備部門都并沒有受到監(jiān)管;直到2006年,印度中央藥品標(biāo)準(zhǔn)控制組織(CDSCO)通知了15類醫(yī)療設(shè)備需要進行注冊,這樣的時代才算宣告結(jié)束。
為了配合落實“印度制造”計劃,CDSCO發(fā)布了新的《醫(yī)療器械法規(guī)2017》(Medical Device Rules,2017),該法規(guī)于2018年1月1日正式開始實施。在實施《醫(yī)療器械法規(guī)2017》之前,根據(jù)《藥品和化妝品法案1940》(Drug and Cosmetic Act, 1940),在印度醫(yī)療器械都是按照藥品標(biāo)準(zhǔn)受到監(jiān)管的。因此,有必要將藥品和醫(yī)療器械區(qū)分開來。其次,也迫切需要為當(dāng)?shù)刂圃焐淘谟《劝l(fā)展行業(yè)提供更有利的環(huán)境。最后,工商部在2017年發(fā)布了公共采購方案,并將藥品部門確定為公告機構(gòu)。
制定新法規(guī)是為了促進國內(nèi)制造業(yè)的發(fā)展并且規(guī)范該地區(qū)的進口和制造業(yè)。目前,跨國公司占據(jù)了印度醫(yī)療器械市場75%的銷售額。新法規(guī)遵循了GHTF(全球協(xié)調(diào)工作組)的指南并且與其中基于風(fēng)險的分類方法一致。此外,公告機構(gòu)的檢查也被引入到了新的醫(yī)療器械法規(guī)中來。本文強調(diào)了一些要點以便于更好地理解《醫(yī)療器械法規(guī)2017》。
分類
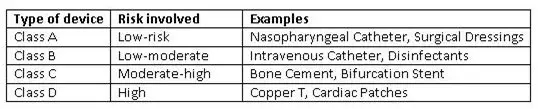
與全球法規(guī)一致,新法規(guī)引入了基于風(fēng)險的分類系統(tǒng)。CDSCO將醫(yī)療設(shè)備進行了分類,并在其網(wǎng)站上不時發(fā)布分類的設(shè)備列表。進口商和制造商必須按照分類表對他們的設(shè)備進行分類。如果一個設(shè)備在GHFT國家的分類等級更高,那么將對其考慮更高級別的分類。
《醫(yī)療器械法規(guī)2017》中的設(shè)備分類系統(tǒng)
質(zhì)量管理體系(QMS)評估
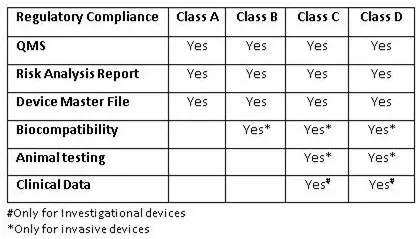
根據(jù)新法規(guī),一種經(jīng)過公告機構(gòu)“第三方合格評定和認(rèn)證”新程序被引入。公告機構(gòu)可以在生產(chǎn)場所對A類和B類設(shè)備執(zhí)行QMS評估。根據(jù)要求,公告機構(gòu)也可以支持CDSCO為C類和D類醫(yī)療設(shè)備的生產(chǎn)現(xiàn)場進行QMS評估。經(jīng)認(rèn)證的機構(gòu)名單將會在其網(wǎng)站上公示。對于國外制造商,CDSCO也可能會要求由CDSCO內(nèi)部檢查員或者任何公告機構(gòu)對其海外生產(chǎn)基地進行檢查。
注冊
新法規(guī)將強制要求所有設(shè)備獲得制造和進口許可證。所有制造和進口許可證的申請都是通過在線門戶網(wǎng)站SUGAM來處理的,SUGAM是一個隸屬于印度衛(wèi)生和家庭福利部(Ministry of Health and Family Welfare)的在線授權(quán)系統(tǒng)。
印度國家授權(quán)許可機構(gòu)(SLA)將對A類和B類設(shè)備的制造許可進行監(jiān)管,而C類和D類許可申請將提交給中央授權(quán)許可機構(gòu)(FSSAI)。質(zhì)量評估報告(QAR)必須與B類、C類和D類設(shè)備的制造許可證申請一同提交。而A類醫(yī)療設(shè)備的QAR只需要在制造許可證頒發(fā)之日起120天內(nèi)提交即可。
Requirements for approval of product
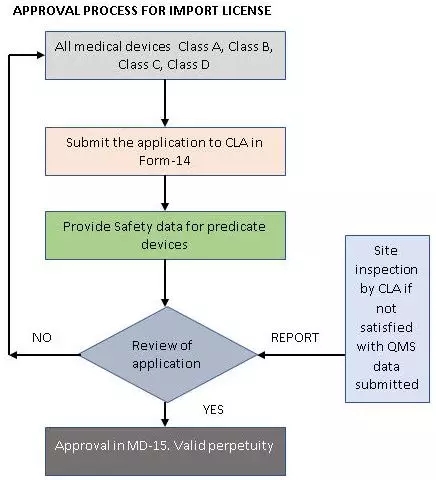
要獲得進口許可證,需要先獲得制造或分銷許可證。國外制造商應(yīng)指定一名獲得授權(quán)的印度代理商來持有許可證,才能進行上市后監(jiān)察(post marketing surveillance,PMS)活動和醫(yī)療設(shè)備的分銷。所有類別的醫(yī)療器械進口許可證申請都必須提交中央授權(quán)許可機構(gòu)。
與制造許可證不同的是,進口許可證的申請不需要進行QAR,但如果需要,中央授權(quán)許可機構(gòu)可能會檢查國外的場地。為了保證新的指導(dǎo)方針更加嚴(yán)格,進口商現(xiàn)在必須向中央授權(quán)許可機構(gòu)提交完整的技術(shù)文件和進口許可證申請,并且每個印度代理商將負(fù)責(zé)在國內(nèi)的PMS活動。根據(jù)2017年的新法規(guī),相同產(chǎn)品的多個進口許可證可以由不同的印度代理商代理。
所有獲得授予的許可都是永久性的,除非它們被取消。為了保留許可證,持有人每五年需要支付一次執(zhí)照留置費。印度政府特別注意到了時間表是不可避免的,因此合理化設(shè)置了授予許可證的時間。
獲得醫(yī)療器械制造/進口許可證的時間
臨床研究
《醫(yī)療器械法規(guī)2017》改變了醫(yī)療器械臨床試驗方案,將其從一個與藥物相同的四期試驗改成了兩期試驗。這兩期將分為實驗性臨床研究(探索性研究)和關(guān)鍵臨床研究(驗證性研究)。除此之外,PMS還必須在獲得市場批準(zhǔn)后才能進行。然而,對于一種在印度還沒有已獲批準(zhǔn)類似器械的醫(yī)療器械申請進口許可證,如果已經(jīng)獲得澳大利亞、加拿大、日本、美國或歐盟成員國機構(gòu)當(dāng)局頒發(fā)的自由銷售證書(FSC),就不需要進行臨床研究。
此外,在新法規(guī)中引入了用于醫(yī)療器械(除科研型醫(yī)療器械)已獲批準(zhǔn)類似器械的“實質(zhì)等同性原則(Substantial Equivalence)”。對于體外診斷產(chǎn)品,“臨床表現(xiàn)評估”將成為監(jiān)管要求的一部分。此外,臨床試驗所產(chǎn)生的數(shù)據(jù)如果不是用于申請制造或進口許可證的話,將不需要事先獲得臨床試驗批準(zhǔn)。
標(biāo)簽
根據(jù)新法規(guī)中的規(guī)格說明,標(biāo)簽要求被強制性要求遵守。除此之外,印度政府還強制執(zhí)行了《法定計量(包裝商品)法規(guī)2011》。此外,每個設(shè)備的醫(yī)療器械唯一標(biāo)識(UDI)號碼必須在標(biāo)簽上注明,有效期至2022年1月。
召回
藥品和化妝品法案不能強制制造商或進口商從市場上撤回產(chǎn)品。而根據(jù)新法規(guī),制造商或進口商必須召回任何危險或有害的產(chǎn)品,并要求他們提供召回的理由。
公共采購訂單(PPO)
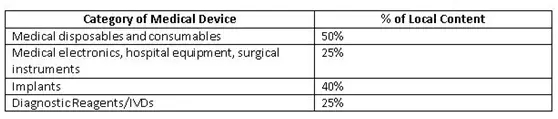
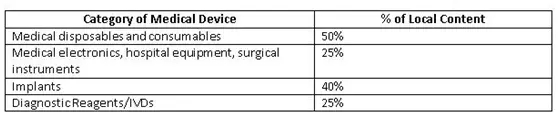
為了改善國內(nèi)制造商生態(tài)系統(tǒng),印度政府(GoI)為公共采購訂單(PPO)制定了準(zhǔn)則草案。這些規(guī)定覆蓋了價值50 lakh或以下的公共采購醫(yī)療設(shè)備的標(biāo)書。當(dāng)?shù)爻煞郑╨ocal content)成本的確定應(yīng)以人力和材料(主要成分)的原產(chǎn)國為基礎(chǔ)。政府提出了一個計算當(dāng)?shù)爻煞值墓剑?
D =(A / C)* 100
?。– = A + B)
D=當(dāng)?shù)爻煞值陌俜直?
C =總成本
B=進口部件成本
A=國產(chǎn)部件成本
Minimum local content in domestic medical devices fixed by GoI
結(jié)論
《醫(yī)療器械法規(guī)2017》有著許多能夠鼓勵印度醫(yī)療器械行業(yè)發(fā)展的特點。通過引入單個在線門戶,注冊流程已經(jīng)被簡化。公告機構(gòu)的審計將進一步提高設(shè)備制造的質(zhì)量。臨床試驗要求的改變將鼓勵新醫(yī)療設(shè)備的創(chuàng)新。這些規(guī)定將鼓勵國產(chǎn)制造,并加強對進口許可證文件的審查。